


Vol. XXXIV Issue 2 – December 2023
Article 1
DOI: 10.35407/bag.2023.34.02.01
ARTÍCULOS ORIGINALES
ALT-EJ GENERA REARREGLOS CROMOSÓMICOS EN RESPUESTA A ETOPÓSIDO EN CÉLULAS HUMANAS CON LOS PRINCIPALES SISTEMAS DE REPARACIÓN DE RUPTURAS DE DOBLE CADENA COMPROMETIDOS
ALT-EJ ORIGINATES CHROMOSOMAL REARRANGEMENTS IN RESPONSE TO ETOPOSIDE IN HUMAN CELLS WITH THE MAIN DNA DOUBLE-STRAND BREAK REPAIR SYSTEMS COMPROMISED
Kramar J.1
Palmitelli M.1
De Campos-Nebel M.1
González-Cid M.1*
1 Laboratorio de Mutagénesis, Instituto de Medicina Experimental, IMEX-CONICET, Academia Nacional de Medicina, Buenos Aires, Argentina.
* Corresponding author: Marcela Beatriz González Cid margoncid@gmail.com ORCID 0000-0001-9537-0925
ABSTRACT
The antitumor drug Etoposide (ETO) induces DNA double-strand breaks (DSB) and is associated with the development of secondary neoplasms in treated patients. DSB are repaired by two main mechanisms, homologous recombination (HR) and classical non-homologous end joining (c-NHEJ). When HR and c-NHEJ are defective, DSB are repaired by the PARP-1-dependent alternative end-joining (alt-EJ) pathway. The involvement of alt-EJ in the progression of DSB induced by ETO in the G2 phase of human cells was analyzed. HeLa cells deficient in HR (cohesin RAD21 inhibition, HeLa RAD21kd) and their nonsilencing control (HeLa NS) were established. Cells were treated with ETO in the presence of a chemical inhibitor of DNA-PKcs (DNA-PKi, c-NHEJ). In both cell lines, ETO-induced DSB (γH2AX+) in G2 phase were increased compared to their controls. The incorrect repair of DSB in DNA-PKcs- and RAD21-deficient cells caused a synergistic augment in chromatid exchanges and dicentric chromosomes in the first and second metaphase, respectively. In contrast, the frequency of dicentric chromosomes was reduced in PARP-1-deficient cells (HeLa PARP-1kd) following ETO treatment. In HeLa RAD21kd binucleated cells, DNA-PKi/ETO increased the percentage of cells with ≥20 γH2AX foci in the G1-postmitotic phase and of micronuclei at 96 h. A greater accumulation in G2/M was observed in HeLa NS treated with DNA-PKi/ ETO compared with HeLa RAD21kd at 8 h. The cell cycle restarted in HeLa NS at 16 h; however, the G2/M accumulation was maintained in HeLa RAD21kd. Chromosomal rearrangements obtained when DNAPKcs and RAD21 were absent and their decrease in HeLa PARP-1kd cells suggest that alt-EJ contributes to their formation.
Key words: chromosomal aberrations, cell cycle, cohesin, double-strand breaks, DNA repair pathways.
RESUMEN
La droga antitumoral Etopósido (ETO) induce rupturas de doble cadena en el ADN (RDC) y promueve el desarrollo de neoplasias secundarias en los pacientes tratados. Dos mecanismos principales, recombinación homóloga (HR) y reunión de extremos no-homólogos clásica (c-NHEJ) reparan las RDC. Cuando HR y c-NHEJ son defectuosas, la vía alternativa de reunión de extremos (alt-EJ) dependiente de PARP-1 está implicada. Se examinó la participación de alt-EJ en la progresión de las RDC inducidas por ETO en la fase G2 de células humanas. Se establecieron células HeLa deficientes en HR (inhibición de cohesina RAD21, HeLa RAD21kd) y su control no-silenciada (HeLa NS). Las células se trataron con ETO en presencia del inhibidor químico de DNA-PKcs (DNA-PKi, c-NHEJ). En ambas líneas celulares, la inducción de RDC (γH2AX+) por ETO en la fase G2 aumentó respecto a sus controles. La reparación incorrecta en células deficientes en DNA-PKcs y RAD21 originó un incremento sinérgico de intercambio de cromátidas y de cromosomas dicéntricos en la primera y segunda metafase, respectivamente. En cambio, la frecuencia de cromosomas dicéntricos se redujo en células deficientes en PARP-1 (HeLa PARP1kd) luego del tratamiento con ETO. En células binucleadas HeLa RAD21kd, DNA-PKi/ETO acrecentó el porcentaje de células con ≥20 focos γH2AX en la fase G1-posmitótica y de micronúcleos a las 96 h. Una mayor acumulación en G2/M se observó en HeLa NS tratadas con DNA-PKi/ETO en relación a HeLa RAD21kd a las 8 h. El ciclo celular se reanudó en HeLa NS a las 16 h, sin embargo, la acumulación se mantuvo en HeLa RAD21kd. Los rearreglos cromosómicos obtenidos con DNA-PKcs y RAD21 disfuncionales y su disminución en células HeLa PARP-1kd, sugieren que alt-EJ contribuye a su formación.
Palabras Claves: aberraciones cromosómicas, ciclo celular, cohesina, rupturas de doble cadena, vías de reparación del ADN.
General Editor: Elsa Camadro
Recived 03/19/2022
Revised version received: 04/21/2022
Accepted: 07/05/2022
INTRODUCCIÓN
Las rupturas de doble cadena (RDC) en el ADN son consideradas lesiones perjudiciales para las células debido a que su falta de reparación o reparación incorrecta pueden llevar a mutaciones, pérdida de información genética o rearreglos cromosómicos y así, promover un proceso tumorigénico (Pierce et al., 2001). El hecho que las RDC sean lesiones citotóxicas es explotado en el tratamiento convencional del cáncer empleando drogas quimioterapéuticas inductoras de este tipo de daño, como es el caso de la epipodofilotoxina etopósido (ETO) (Kopa et al., 2019). Esta droga actúa como veneno de ADNtopoisomerasa II (Top2), pues convierte a esta enzima en una potente toxina que fragmenta al genoma en su presencia (Baldwin y Osheroff, 2005). Las enzimas Top2 resuelven problemas topológicos del ADN durante la replicación, transcripción, segregación y condensación cromosómica catalizando la liberación de torsiones sobre el mismo mediante la introducción de rupturas transitorias en la doble hélice (McKie et al., 2021). ETO se caracteriza por estabilizar los complejos entre el ADN y Top2, conocidos como complejos de clivaje, previniendo la religación de los extremos rotos y conduciendo a la formación de RDC persistentes (Montecucco et al., 2015).
Los mecanismos de reparación de las RDC en células humanas son generalmente agrupados en tres categorías, dependiendo del uso o no de una secuencia de ADN como templado (Ranjha et al., 2018; Scully et al., 2019; Vítor et al., 2020): la vía de recombinación homóloga (homologous recombination, HR), la vía clásica de reunión de extremos no homólogos (classical nonhomologous end joining, c-NHEJ) y la vía alternativa de reunión de extremos (alternative end-joining, alt-EJ). HR actúa en forma lenta, y es un mecanismo preciso debido a que utiliza una molécula de ADN sin daño, la cromátida hermana, como templado para restituir la información original. En el proceso de reparación mediante c-NHEJ se produce la ligación de los extremos rotos del ADN sin necesidad de una secuencia homóloga. La característica más destacada de esta vía es la velocidad con la cual funciona suprimiendo en gran medida, la formación de translocaciones cromosómicas; no obstante, pueden surgir alteraciones en la secuencia del ADN en los sitios de unión. En el caso de alt-EJ, la vía opera con velocidad y fidelidad marcadamente más bajas que c-NHEJ, provocando translocaciones cromosómicas con mayor frecuencia y generando, en el sitio de unión, alteraciones más extensas en la secuencia del ADN. Un subgrupo de alt-EJ emplea secuencias de microhomología a cada lado de la RDC para usar como templado, luego estas secuencias son removidas y se produce la síntesis del ADN y la unión de los extremos rotos (Wang y Xu, 2017).
La contribución relativa de estas vías depende de la fase del ciclo celular donde se haya producido la RDC. Mientras que HR actúa durante las fases S tardía y G2 donde se realiza la síntesis del ADN y se dispone de una cromátida hermana como templado, la vía c-NHEJ opera a lo largo del ciclo celular, actuando principalmente en las fases G0/G1 y G2. Por último, alt-EJ permanece activa a lo largo del ciclo celular, mostrando un marcado aumento durante la fase G2 (Mladenov et al., 2016).
La regulación de la vía HR está limitada por el complejo multiproteico cohesina (Watrin y Peters, 2006). En organismos multicelulares, este complejo está compuesto por RAD21, las subunidades del mantenimiento estructural de los cromosomas SMC1 y SMC3, STAG1 (SA1) y STAG2 (SA2) (Peters et al., 2008). Luego de la formación de una RDC, la cohesina se acumula sobre la cromatina rodeando el sitio de la ruptura para una eficiente reparación (Watrin y Peters, 2006). La estabilización de la cohesina en las RDC depende de la subunidad RAD21 (Heidinger-Pauli et al., 2009). La proteína RAD21 (llamada también MCD1 o SCC1) está involucrada en la cohesión de las cromátidas hermanas desde la replicación del ADN en la fase S hasta la segregación apropiada de los cromosomas durante mitosis y se conserva evolutivamente desde las levaduras hasta los seres humanos. La expresión del ARNm de RAD21 está regulada durante el ciclo celular, aumentando en la fase S y alcanzando su máxima expresión en G2 (Cheng et al., 2020). Además de ser requerida para el proceso de HR, RAD21 participa en el punto de control (checkpoint) G2/M en respuesta al daño inducido en el ADN (Litwin et al., 2018).
La proteína quinasa dependiente de ADN (DNA-PK) participa en la reparación de las RDC a través de c-NHEJ y consiste de una subunidad catalítica (DNA-PKcs) y de un heterodímero compuesto por las subunidades KU70/KU80 (Mohiuddin y Kang, 2019). La formación de este complejo quinasa activo causa la fosforilación de proteínas implicadas en esta vía, como así también la autofosforilación de DNA-PKcs. Ambos procesos influyen en la estabilización de la unión entre DNA-PKcs y los extremos del ADN.
Por último, la vía alt-EJ depende de la polimerasa poli-(ADP-ribosa)-1, PARP-1, la cual tiene afinidad por unirse a los extremos de ADN, alterar la estructura de la cromatina y atraer proteínas de reparación al sitio de daño (De Vos et al., 2012). Esta vía contribuye escasamente en la reparación de las RDC en células normales (no malignas), sin embargo, existe un interés creciente en esta vía porque genera reordenamientos genómicos que son característicos de las células tumorales (Sallmyr y Tomkinson, 2018). En base a esto, se sugiere que alt-EJ no es una vía de reparación de las RDC funcionalmente optimizada y específica, sino un intento de último recurso de la célula para eliminar los extremos de ADN desprotegidos empleando actividades enzimáticas preexistentes cuando los dos mecanismos principales están afectados (Iliakis et al., 2015). Así, en un comienzo se estableció que alt-EJ funcionaba como respaldo (backup) de c-NHEJ; sin embargo, estudios posteriores demostraron que este mecanismo puede también participar en la reparación de las RDC cuando la vía HR no está presente (Ceccaldi et al., 2016).
En presencia del ADN dañado, los puntos de control evitan la progresión del ciclo celular, dando tiempo para que la reparación ocurra y se eviten alteraciones cromosómicas capaces de manifestarse en las generaciones posteriores; y en el caso de la existencia de lesiones irreparables, se estimule la muerte celular. Los defectos en los puntos de control aumentan la inestabilidad genómica y contribuyen al desarrollo tumoral (Visconti et al., 2016).
Nuestro objetivo fue analizar la participación de la vía alt-EJ y del punto de control G2/M en la progresión del daño en el ADN de células humanas tratadas con ETO en la fase G2 del ciclo celular, y establecer la posible contribución de alt-EJ en la inducción de rearreglos cromosómicos típicos de las neoplasias secundarias asociadas al tratamiento con venenos de Top2.
MATERIALES Y MÉTODOS
Agentes químicos
ETO (Sigma-Aldrich), NU7026 (Calbiochem), inhibidor de DNA-PKcs (DNA-PKi) y citocalasina B (Calbiochem) se disolvieron en dimetilsulfóxido (DMSO). Puromicina (Sigma-Aldrich) se disolvió en agua bidestilada.
Cultivos celulares y silenciamiento génico de RAD21 y PARP-1
Células humanas HeLa (carcinoma cérvico-uterino) se cultivaron en medio completo conteniendo RPMI 1640 con 10% de suero fetal bovino, 2 mM de L-glutamina y antibióticos (100 U/ml de penicilina y 100 µg/ml de estreptomicina). Los cultivos se mantuvieron a 37º C en una atmósfera húmeda con 5% CO2. Las células se transfectaron con el plásmido pGIPZ conteniendo el shRNAmir (microRNA-adapted short hairpin RNA) específico contra el ARNm de los genes humanos RAD21 (Open Biosystems, clone id: V2LMM_16122) o PARP-1 (Open Biosystems, clone id: V2LHS_201984) para realizar su silenciamiento (knock-down). Se generaron las líneas HeLa RAD21kd y HeLa PARP1kd, y como control negativo se utilizó un plásmido pGIPZ codificando una secuencia No-Silenciante (Open Biosystems, #RHS4346, HeLa NS). Las transfecciones se realizaron usando Lipofectamina 2000 (Invitrogen) de acuerdo a las instrucciones del fabricante. A las 48 h, las células se crecieron en medio completo en presencia de puromicina 1 µg/ml, con renovación de este medio cada tres días. El proceso de selección se realizó durante 3-4 semanas y se establecieron clones. El nivel de silenciamiento de estos clones se evaluó mediante la retrotranscripción asociada a la reacción en cadena de la polimerasa en tiempo real (qRT-PCR).
Técnica qRT-PCR
Se extrajo el ARN de las células HeLa deficientes en RAD21 o en PARP-1 y de las células HeLa NS con el reactivo TRIReagent (Molecular Reagent Center) y se determinó su concentración por espectrofotometría. El ARN total (1 g/reacción) se retrotranscribió usando cebadores (primers) al azar (Biodynamics) y la enzima M-MLV (Promega). El análisis de PCR en tiempo real se realizó con “Mezcla Real” (Biodynamics) y la reacción se llevó a cabo en un termociclador CFX Connect (BioRad). Los primers para amplificar los genes humanos
RAD21 (FW: 5’-GGATAAGAAGCTAACCAAAGCCC-3’, RV: 5’-CTCCCAGTAAGAGATGTCCTGAT-3’), PARP-1 (FW: 5’-CGGAGTCTTCGGATAAGCTCT-3´, RV: 5’-TTTCCATCAAACATGGGCGAC-3´) y β-2-Microglobulina (B2M) (FW: 5’-AAGATGAGTATGCCTGCCGTGTGA-3’, RV: 5’-ACCTCTAAGTTGCCAGCCCTCCTA-3’) se adquirieron en Genbiotech, siendo B2M el gen usado como referencia. Los porcentajes de reducción de los ARNm de RAD21 y de PARP-1 se estimaron en relación a la expresión relativa con B2M de acuerdo al método de Pfaffl (2004) en dos experimentos independientes.
Intercambio de Cromátidas Hermanas (ICH) en células metafásicas
Las células HeLa NS y HeLa RAD21kd se sembraron en placas de 60 mm y se cultivaron en medio completo en presencia de 10 μg/ml de la base análoga de timidina, bromodeoxiuridina (BrdU). A las 40 h (dos ciclos de replicación), las células se expusieron a Colcemid 0,1 μg/ ml durante los últimos 90 min de cultivo, luego se realizó la hipotonía con una solución de KCl 0,075 M y se fijaron en metanol: ácido acético glacial (3:1) para la obtención de metafases. Los extendidos cromosómicos se dejaron envejecer por lo menos durante una semana. Luego se colocaron en una solución de Hoechst 33258 1 μg/ml en agua destilada por 15 min en oscuridad, se activaron por exposición a la luz solar por 3-4 h y se incubaron en 2X SSC en baño a 56º C por 1 h. Finalmente, se colorearon con Giemsa 10% en buffer Sorensen durante 4 min. Se contaron los ICH/cromosoma en 25 metafases en dos experimentos independientes.
Ensayo clonogénico
Células HeLa NS y HeLa RAD21kd (8x102/placa) se sembraron en placas de 60 mm y trataron con concentraciones crecientes de ETO (0,05; 0,1; 0,5 y 1 µg/ ml) durante 16 h. Las células se lavaron con solución de buffer fosfato (PBS) y se cultivaron en medio completo por 14 días. Luego, las células se fijaron con metanol 100% y se colorearon con violeta de cristal 1%. Se contaron las colonias conteniendo >50 células y la eficiencia de plaqueo se determinó como (número de colonias contadas/número de células sembradas) x 100, en los cultivos control sin tratamiento. La fracción de sobrevida se estimó como la relación de colonias en cultivos tratados en relación con los cultivos controles y se expresó en porcentaje. Se realizaron tres-cuatro experimentos independientes.
Determinación de la concentración de ETO a ser utilizada mediante la evaluación de la progresión del ciclo celular
Células HeLa NS y HeLa RAD21kd se sembraron en placas de seis pocillos y se trataron con concentraciones crecientes de ETO (1; 2; 5 y 10 µg/ml) durante 2 h. Las células control se expusieron a DMSO 0,5% durante 2 h. Las células se lavaron con PBS, se cultivaron en medio completo durante 24 h y se cosecharon las células unidas al sustrato y aquellas presentes en el medio completo. Luego se fijaron en metanol 90% a -20º C durante al menos 2 h. Se lavaron en PBS y se resuspendieron en 300 μl de PBS conteniendo 3 μl de RNAsa A (libre de DNAsas, 200 µg/ml) y 8 μl de Ioduro de Propidio (IP, 10 µg/ml) durante un mínimo de 20 min en oscuridad, a temperatura ambiente. La adquisición de 20.000 células por concentración se realizó mediante un citómetro de flujo FACSCalibur (Becton-Dickinson). El análisis del ciclo celular se realizó utilizando el software FSC Express4. Se determinó el porcentaje de células presentes en la fase G2/M en tres experimentos independientes.
Detección de RDC (γH2AX) inducidas por ETO en la fase G2
Células HeLa NS y HeLa RAD21kd se sembraron en placas de 35 mm y se trataron con DMSO 0,5% o con ETO 2 μg/ml. A las 2 h se recogió el medio completo y se tripsinizaron y cosecharon las células. Luego de la centrifugación, las células se lavaron con PBS y se fijaron en metanol 90% a -20º C durante al menos 2 h. Las células se marcaron con el anticuerpo primario anti-γH2AX (1:200, Millipore clone JBW301) seguido con el anticuerpo secundario conjugado con Alexa Fluor 488 (1:250). Luego se trataron con RNAsa A e IP. La adquisición de 20.000 células por tratamiento se realizó mediante el citómetro de flujo FACSCalibur usando el software Cell Quest. Se determinó el porcentaje de células γH2AX+ en la fase G2 en cuatro experimentos independientes.
Aberraciones cromosómicas estructurales
Células HeLa NS y HeLa RAD21kd se sembraron en placas de 60 mm y se trataron con DMSO 0,5% durante 2 h, con el inhibidor de DNA-PKcs (DNA-PKi) NU7026 10 µM, durante todo el tiempo de cultivo, con ETO 2 µg/ml por 1 h, o una combinación de ambas drogas (pretratamiento con DNA-PKi durante 1 h, tratamiento con ETO durante 1 h y tratamiento con DNA-PKi durante el resto del tiempo de cultivo). Las células permanecieron en medio completo hasta alcanzar la primera (5-6 h) o la segunda (28 h) metafase postratamiento. A su vez, las células HeLa PARP-1kd se trataron con DMSO 0,5% durante 2 h o con ETO 2 µg/ml por 1 h y se mantuvieron en medio completo hasta la segunda metafase. Las células HeLa NS, RAD21kd y PARP-1kd se sometieron al procesado citogenético convencional: Colcemid, solución hipotónica con KCl 0,075 M, fijación en metanol: ácido acético glacial (3:1) y coloración con Giemsa 10% durante 4 min. Se evaluó la presencia de aberraciones cromosómicas de tipo cromatídico o cromosómico en un mínimo de 50 metafases por tratamiento. Además, a las 28 h postratamiento, se analizó el índice mitótico (IM) contando el número de metafases en 1.000 núcleos interfásicos y se expresó en porcentajes. El análisis se realizó mediante microscopio óptico en dos-cinco experimentos independientes.
Análisis del ciclo celular
Células HeLa NS y HeLa RAD21kd se sembraron en placas de 35 mm y se trataron con DMSO 0,5% durante 2 h, con DNA-PKi durante todo el tiempo de cultivo, con ETO 2 µg/ml por 1 h o una combinación DNA-PKi/ETO (pretratamiento con NU7026 durante 1 h, tratamiento con ETO durante 1 h y tratamiento con NU7026 durante el resto del tiempo de cultivo). Las células se dejaron en medio completo durante 8 y 16 h postratamiento. Se siguió el procedimiento descripto en Determinación de la concentración de ETO a ser utilizada. Las fases del ciclo celular se analizaron mediante el software FSC Express4 en cuatro experimentos independientes.
Focos de γH2AX en los núcleos de células binucleadas
Células HeLa NS y HeLa RAD21kd se sembraron sobre cubreobjetos en placas de 35 mm y se trataron con DMSO, DNA-PKi, ETO o una combinación de DNA-PKi/ ETO y se mantuvieron en medio completo durante 10-11 h. Se agregó citocalasina B 3 μg/ml durante las últimas 4 h de cultivo. Las células se expusieron a la solución hipotónica KCl 0,075 M durante 8 min, se fijaron con paraformaldehído (PFA) 2% y se permeabilizaron con Tritón X-100 0,25%. Se realizó la marcación usando el anticuerpo primario anti-γH2AX (1:200) y el anticuerpo secundario conjugado con Alexa Fluor 594 (1:300). El ADN se coloreó con DAPI (4,6-diamidino-2-fenilindol) y los cubreobjetos se colocaron sobre portaobjetos. En estos preparados se contaron los núcleos interfásicos que contenían ≥20 focos γH2AX en 500 células binucleadas (BN) por tratamiento. Además, se evaluó el porcentaje de células BN en 1.000 células por tratamiento. Se realizaron dos-cuatro experimentos independientes.
Micronúcleos en células binucleadas en combinación con γH2AX
Células HeLa NS y HeLa RAD21kd se sembraron y trataron según lo descrito en el punto anterior, manteniéndose en medio completo durante 96 h. Se adicionó citocalasina B 3 µg/ml durante las últimas 12 h de cultivo y las células se expusieron a solución hipotónica, fijación con PFA y permeabilización con Tritón X-100. La inmunomarcación se efectuó usando el anti-γH2AX y el anticuerpo secundario conjugado con Alexa Fluor 594. El ADN se coloreó con DAPI. Los micronúcleos (MN) y la presencia o no de focos γH2AX en los MN se contaron en 400 células BN por tratamiento. Se realizaron dos-tres experimentos independientes.
Análisis estadístico
Los datos se presentaron como media ± error estándar de la media y el análisis estadístico se realizó mediante el test t de Student para evaluar la significancia entre los tratamientos en las líneas celulares empleadas usando el software GraphPad Prism 8.0 (GraphPad Software Inc., USA). La relación lineal dosis-respuesta se analizó mediante el coeficiente de correlación de Pearson (Primer of Biostatistics, Stanton A. Glanty, by McGrawHill, Inc, version 3.0, 1992). La significancia estadística se consideró cuando p<0,05.
RESULTADOS
El nivel de expresión del gen RAD21 en las líneas celulares generadas se evaluó mediante qRT-PCR (Figura 1a). Los porcentajes de expresión en las líneas celulares silenciadas fueron: HeLa shRAD21#1= 32,2±5,8%; HeLa shRAD21#2= 38,4±9,3% y HeLa shRAD21#3=54,2±5,4% en relación a la línea control HeLa NS (100%). No existieron diferencias estadísticamente significativas en los niveles de expresión de RAD21 entre los clones generados. Se decidió utilizar como modelo experimental las células del clon HeLa shRAD21#1, a partir de ahora llamadas células HeLa RAD21kd, por presentar el nivel de expresión más bajo.
En primer lugar, se caracterizaron las células HeLa RAD21kd generadas. La subunidad del complejo cohesina RAD21 mantiene las cromátidas hermanas unidas en el sitio de las RDC para permitir la reparación mediante HR y esta vía está asociada a la presencia de ICH en células de vertebrados. Por lo tanto, se examinó la frecuencia espontánea de ICH/cromosoma en las células HeLa RAD21kd (Figura 1b). Los resultados mostraron valores menores en las células deficientes en RAD21 que aquellos obtenidos en las células proficientes HeLa NS (0,09±0,01 vs. 0,13±0,01, p=0,0001).
Para determinar la sensibilidad de la línea celular HeLa RAD21kd, las células se expusieron a concentraciones crecientes de ETO durante 16 h (Figura 1c) y se evaluó la formación de colonias sobrevivientes a los 14 días. La eficiencia de plaqueo en células controles sin tratar se usó para normalizar el número de colonias en HeLa NS y RAD21kd. La eficiencia fue de 81,4±3,2% para HeLa NS y de 68,6±2,9% para HeLa RAD21kd. Ambas líneas celulares mostraron una relación lineal entre las colonias formadas y las concentraciones utilizadas de ETO (HeLa NS r= 0,986, p=0,002 y HeLa RAD21kd r=0,903, p=0,036). Las células HeLa RAD21kd presentaron una mayor sensibilidad a ETO a partir de 0,05 µg/ml (p<0,02).
Luego se estableció la concentración de ETO a ser utilizada en los siguientes experimentos mediante el análisis de la progresión del ciclo celular. Las células HeLa NS y HeLa RAD21kd se expusieron a ETO desde 1 a 10 µg/ml durante 2 h y se determinó el porcentaje de células en la fase G2/M a las 24 h (Figura 1d). Los datos obtenidos indicaron una mayor acumulación de células en la fase G2/M a medida que la concentración de ETO aumentaba (HeLa NS r= 0,993, p=0,0001 y HeLa RAD21kd r=0,980, p=0,003). El arresto en G2/M con ETO 2 µg/ml fue de 24,4±3,2% para HeLa NS y de 31,0±4,4% para HeLa RAD21kd, sin existir diferencias significativas entre ambos valores. A partir de esta concentración, el porcentaje de células arrestadas en G2/M fue del 50% para ETO 5 µg/ml y superior al 65% para ETO 10 µg/ml en las dos líneas celulares, produciendo una importante acumulación en esta fase. En base a estos resultados, se eligió la concentración subtóxica de 2 µg/ml.
Por último, se analizó la formación de RDC mediante el biomarcador γH2AX en la fase G2 de las células Hela NS y RAD21kd (Figura 1e). La inducción de γH2AX es uno de los primeros eventos detectados en las células luego de la exposición a agentes que dañan al ADN. El porcentaje de células HeLa NS γH2AX+ tratadas con ETO fue de 88,2±2,0% y en las células HeLa RAD21kd de 92,2±1,9% en relación a los controles tratados con DMSO, 13,0±1,5% y 14,7±3,3%, respectivamente (p=0,0001). Este resultado permitió establecer que la inducción de RDC por ETO en la fase G2 fue similar en ambas líneas celulares.
Los estudios se realizaron tratando las células en la fase G2 debido a que en esta fase la isoforma α de Top2, blanco específico de ETO, alcanza su máxima expresión (Chen et al., 2015) y los mecanismos de reparación c-NHEJ, alt-EJ y HR se encuentran todos funcionales en dicha fase (Mladenov et al., 2016).
A partir de la elección de la concentración a utilizar, ETO 2 µg/ml, el diseño experimental fue el siguiente: las células HeLa NS y HeLa RAD21kd en crecimiento exponencial, se trataron con DMSO 0,5% por 2 h, o con DNA-PKi 10 µM durante todo el tiempo de cultivo, o con ETO 2 µg/ml por 1 h o con una combinación de ambas drogas, es decir, un pretratamiento con DNA-PKi por 1 h, luego un tratamiento con ETO 2 µg/ml por 1 h, y a continuación de dos lavados con PBS, un tratamiento con DNA-PKi durante el resto del tiempo de cultivo (DNAPKi/ETO). Se analizaron las aberraciones cromosómicas (AC) en la primera (a las 5-6 h) y en la segunda (a las 28 h) metafase, la presencia de focos γH2AX en la fase G1 posmitótica (a las 10-11 h) y en los MN (a las 96 h) de células BN. Además, se evaluó el ciclo celular a las 8 y 16 h (Figuras 2-6).
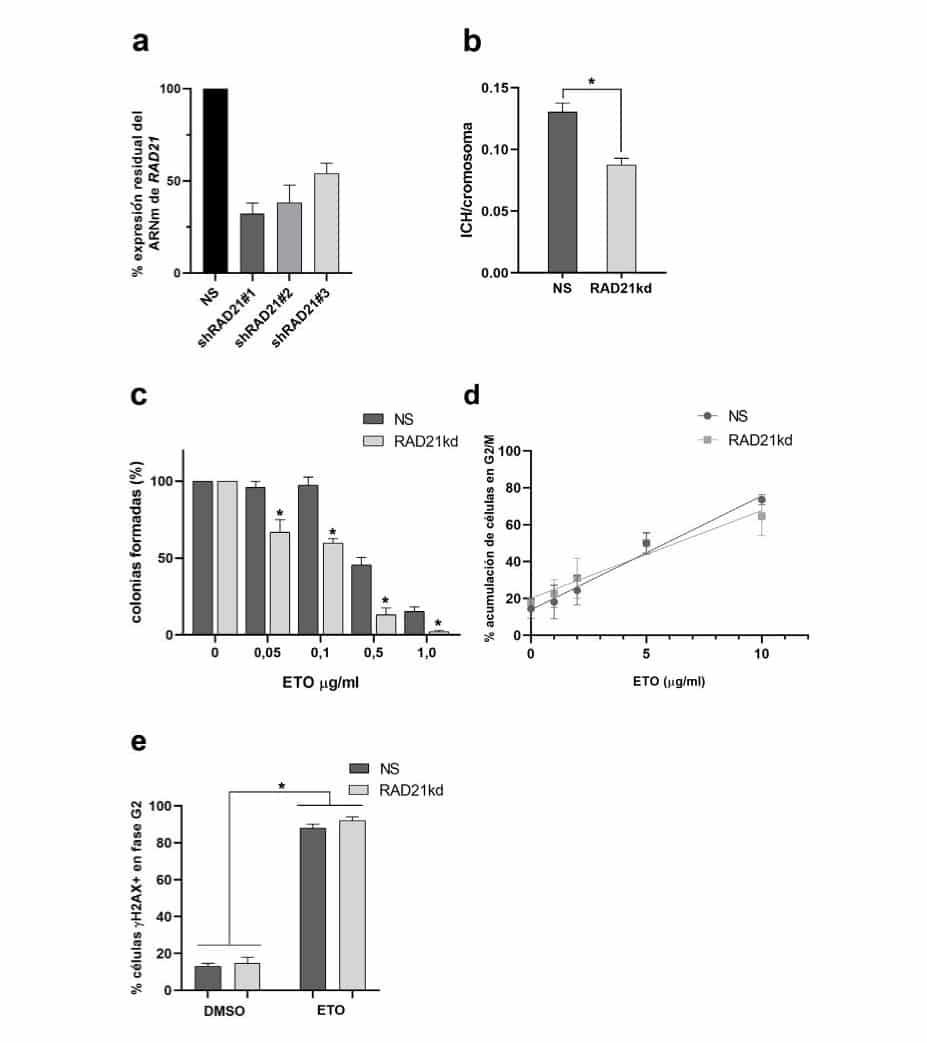
Figura 1. Caracterización de la línea tumoral humana HeLa deficiente en RAD21
a. Porcentaje de expresión de RAD21 en los clones celulares deficientes, HeLa RAD21kd, en relación al control negativo, células HeLa NS (No-Silenciadas); b. Número de intercambios de cromátidas hermanas (ICH) por cromosoma, *p=0,0001; c. Porcentaje de colonias sobrevivientes de células HeLa RAD21kd y HeLa NS tratadas con concentraciones crecientes de ETO (0,05; 0,1; 0,5 y 1 µg/ml) durante 16 h, *p<0,02; d. Porcentaje de células HeLa RAD21kd y HeLa NS acumuladas en el punto de control G2/M luego del tratamiento con ETO 1; 2; 5 y 10 µg/ml durante 2 h; e. Inducción de rupturas de doble cadena (γH2AX+) en la fase G2 de células RAD21kd y Hela NS tratadas con DMSO 0,5% o con ETO 2 µg/ml, durante 2 h, *p=0,0001.
Las RDC en el ADN cuando permanecen sin reparar o son mal reparadas, son lesiones decisivas para la formación de AC (Obe y Durante, 2010). La Figura 2 muestra las AC estructurales en las células HeLa NS y HeLa RAD21kd tratadas con DNA-PKi, con ETO o con la combinación de ambas drogas en la fase G2 y analizadas en la primera metafase, a las 5-6 h posteriores a los tratamientos.
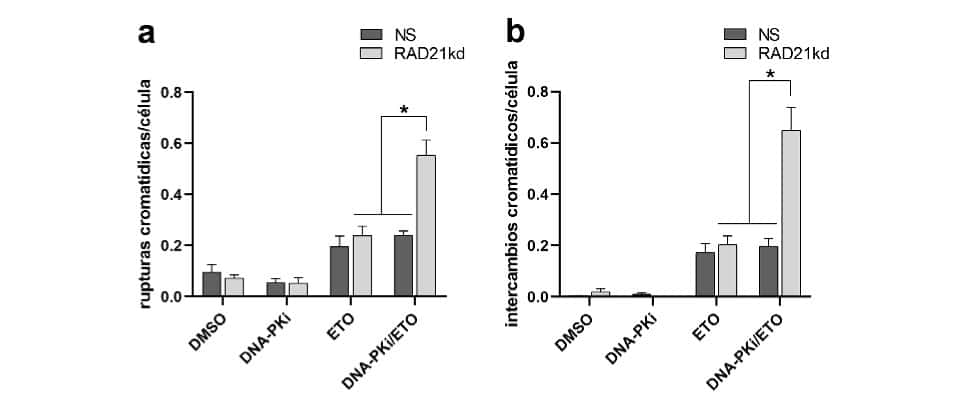
Figura 2. Frecuencia de aberraciones cromosómicas estructurales en la primera metafase (5-6 h) luego de los diferentes tratamientos.
a. Rupturas cromatídicas/célula; b. Intercambios de cromátidas/célula. *p=0,0001.
En las células HeLa NS tratadas con DNA-PKi/ETO se observó una frecuencia de rupturas cromatídicas/ célula de 0,24±0,02 y en las células deficientes en RAD21 tratadas con ETO de 0,24±0,04 (Figura 2a). Las células HeLa RAD21kd expuestas a DNA-PKi/ETO mostraron una frecuencia de 0,55±0,06 (p=0,0001). Cuando se evaluaron las figuras de intercambio de cromátidas/ célula, se observó un fenómeno similar (Figura 2b). Las células HeLa NS tratadas con la combinación de ambas drogas y las células HeLa RAD21kd tratadas con ETO mostraron una frecuencia de 0,20±0,03. En el caso del tratamiento con DNA-PKi/ETO, las células HeLa RAD21kd exhibieron una frecuencia de figuras de intercambio de cromátidas de 0,65±0,09 (p=0,0001).Los resultados obtenidos indicaron que la falta y/o incorrecta reparación de las RDC originaron un aumento sinérgico de rupturas e intercambios de cromátidas frente a ETO en un contexto deficiente en c-NHEJ, por inhibición química de DNA-PKcs y de HR por la deficiencia de RAD21, en comparación a la falta de cada factor en forma individual.
Con el propósito de analizar la progresión del daño a la fase G1 posmitótica, se examinó la presencia de núcleos con ≥20 focos γH2AX en células BN a las 10-11 h (Figura 3). Este estudio se limitó a las células BN que llegaron a la fase G1 resultado del bloqueo de la citocinesis con citocalasina B. Además, el análisis se apoyó en estudios previos de Deckbar et al. (2007), donde fibroblastos humanos irradiados entraban en mitosis sin ser arrestados en el punto de control G2/M, cuando aún tenían hasta ~20 focos γH2AX.
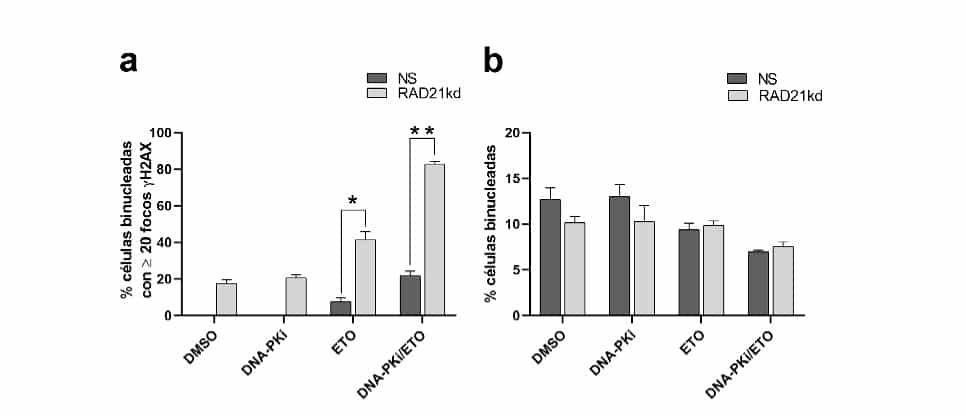
Figura 3. Progresión de las rupturas de doble cadena (γH2AX) a la fase G1 posmitótica (10-11 h) luego de los diferentes tratamientos.
a. Porcentaje de células binucleadas con ≥20 focos γH2AX en Hela RAD21kd y HeLa NS, *p=0,001, **p=0,0001; b. Porcentaje de células binucleadas en HeLa RAD21kd y HeLa NS.
En HeLa RAD21kd, el porcentaje de células BN con ≥20 focos fue ~20% en los controles tratados con DMSO o con DNA-PKi. En HeLa NS no se encontraron células con este número de focos. Los tratamientos con ETO y con DNA-PKi/ETO incrementaron los porcentajes de células BN con ≥20 focos γH2AX, siendo este aumento de 5,4 y 3,8 veces en las células HeLa RAD21kd en comparación con las células HeLa NS, respectivamente (Figura 3a). Sin embargo, en ambas líneas celulares estos tratamientos disminuyeron en forma similar el porcentaje de células BN (Figura 3b).
Los porcentajes observados en las células BN de HeLa NS y HeLa RAD21kd reflejaron una acumulación similar en la fase G2/M a las 8 h. En HeLa NS, luego del tratamiento con ETO fue de 54,8±2,4% y en HeLa RAD21kd, de 52,0±2,9% mientras que, con la combinación DNA-PKi/ETO fue de 60,0±3,0% en HeLa NS y de 59,3±4,7% en HeLa RAD21kd (Figuras 4a y 4b). Sin embargo, el análisis de la proporción de células en G2/M entre aquellas tratadas con DNA-PKi, ETO o DNAPKi/ETO y sus controles tratados con DMSO, mostró que esa proporción fue de 3,9-4,3 en HeLa NS y, en las células HeLa RAD21kd fue de ~2,1 luego de ETO y DNAPKi/ETO. El tratamiento con DNA-PKi en HeLa NS fue de 1,2 y en HeLa RAD21kd fue de 0,95, en relación al control tratado con DMSO.
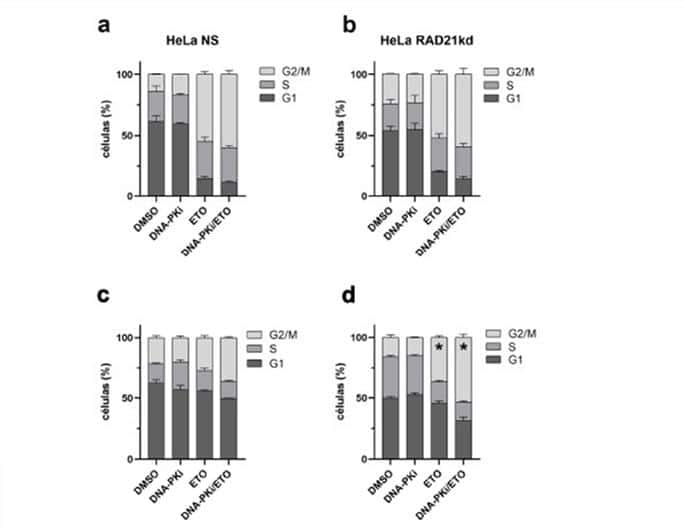
Figura 4. Distribución de las poblaciones celulares en las fases G1, S y G2/M a las 8 h (a y b) y a las 16 h (c y d) luego de los diferentes tratamientos.
*p=0,005, porcentaje de células en la fase G2/M entre ETO vs. DNA-PKi/ETO.
Por lo tanto, la inhibición química de DNA-PKcs y la deficiencia de RAD21 originaron un aumento de AC de tipo cromatídico en la primera metafase y de células BN con RDC (focos γH2AX) persistentes en la fase G1 posmitótica luego del tratamiento con ETO. En cambio, la proporción de células acumuladas en G2/M fue mayor en HeLa NS en comparación con HeLa RAD21kd a las 8 h.
A las 16 h, el porcentaje de células en G2/M fue de ~36%, tanto en las células HeLa NS tratadas con DNA-PKi/ETO como en las células HeLa RAD21kd tratadas con ETO, evidenciando un ingreso al ciclo celular al compararlas con los porcentajes obtenidos a las 8 h (Figura 4c y 4d). A pesar de ello, en HeLa NS, la proporción de células en G2/M entre las tratadas con DNA-PKi/ETO y las tratadas con DMSO fue de 1,6, mientras que en las células HeLa RAD21kd esa proporción fue similar al obtenido a las 8 h (2,2) entre las células tratadas con ETO y aquellas tratadas con DMSO. A su vez, cuando las células HeLa RAD21kd se expusieron a la combinación DNA-PKi/ETO, se observó una acumulación celular en la fase G2/M de 53,3±2,5% (Figura 4d). En consecuencia, la deficiencia de c-NHEJ y HR produjo un arresto en G2/M que iniciado a las 8 h se mantuvo a las 16 h luego del tratamiento con ETO.
En las metafases de la segunda división a las 28 h, las rupturas cromatídicas y cromosómicas/célula, esencialmente fragmentos acéntricos, evidenciaron un aumento aditivo, siendo en las células HeLa RAD21kd tratadas con la combinación DNA-PKi/ETO de 3,3±0,2 (p=0,0001), en células HeLa NS con el mismo tratamiento de 1,8±0,2 y en HeLa RAD21kd tratadas con ETO de 1,3±0,2 (Figura 5a). Cuando se evaluaron los cromosomas dicéntricos/célula en HeLa NS en presencia de DNA-PKi/ ETO y en HeLa RAD21kd expuestas a ETO, el valor fue de ~0,6 (Figura 5b). Con el tratamiento combinado (DNAPKi/ETO) las células HeLa RAD21kd mostraron una frecuencia de 1,90±0,13 (p=0,0001). Ante la ausencia de las vías principales c-NHEJ y HR se produjo un aumento sinérgico de los cromosomas dicéntricos en relación a la frecuencia hallada por la falta individual de actividad de cada proteína, DNA-PKcs y RAD21.
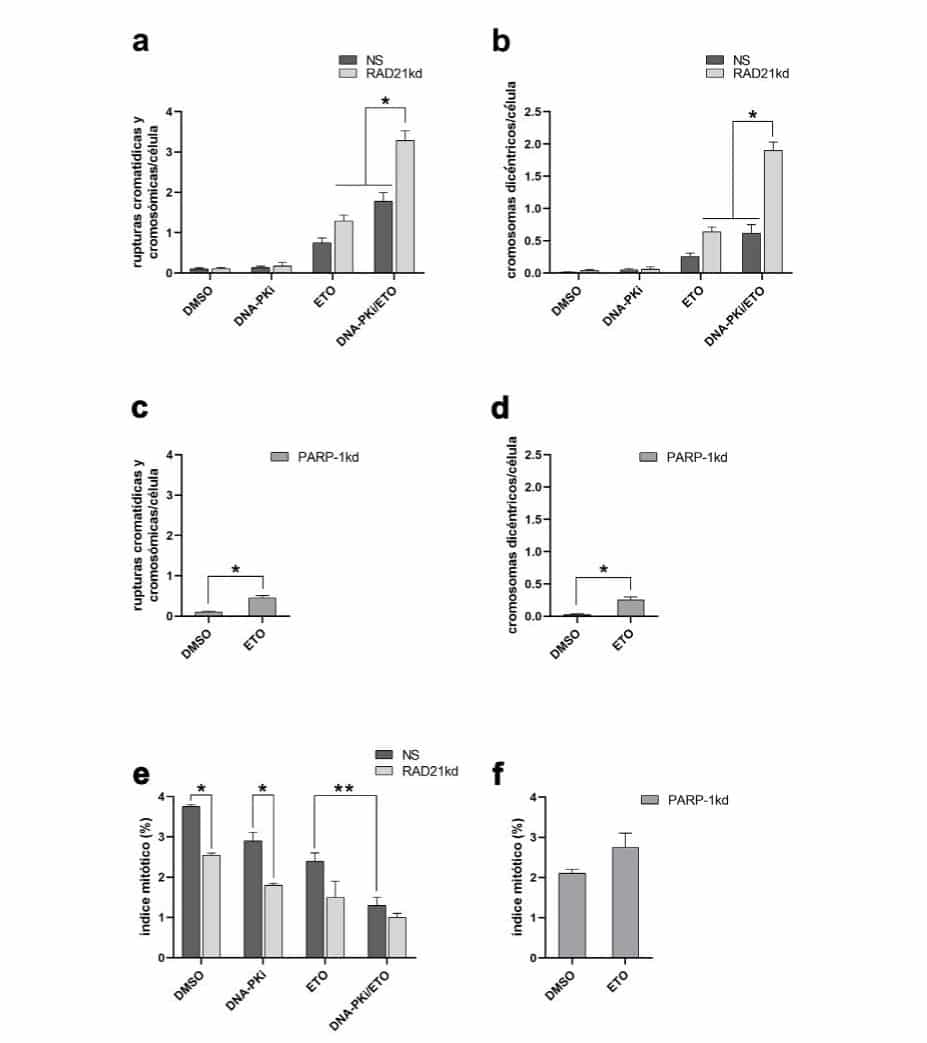
Figura 5. Aberraciones cromosómicas estructurales e índice mitótico en las células HeLa RAD21kd, HeLa PARP-1kd y HeLa NS en la segunda metafase (28 h) luego de los diferentes tratamientos.
a. Rupturas cromatídicas y cromosómicas/célula en células Hela NS y Hela RAD21kd; b. Cromosomas dicéntricos/célula en células Hela NS y Hela RAD21kd, *p=0,0001; c. Rupturas cromatídicas y cromosómicas/célula en células Hela PARP-1kd; d. Cromosomas dicéntricos/célula en células Hela PARP-1kd, *p<0,05; e. Índice mitótico en células HeLa NS y Hela RAD21kd, *p<0,05, **p=0,0001; f. Índice mitótico en células HeLa PARP-1kd.
Como se describió previamente, cuando c-NHEJ y HR no son funcionales, las RDC se reparan por una vía alternativa dependiente de la proteína PARP-1, alt-EJ, la cual origina rearreglos cromosómicos. En base a los resultados obtenidos y para confirmar la participación de alt-EJ en las AC inducidas por ETO, se analizaron las rupturas cromatídicas y cromosómicas y los cromosomas dicéntricos en células deficientes en PARP1 a las 28 h (Figuras 5c y 5d).
Con este propósito, se generó la línea deficiente en PARP-1 y se determinó el nivel de expresión del ARNm del gen PARP-1 mediante la técnica qRT-PCR. La expresión fue de 7,7±4,0% en relación a la línea control HeLa NS (100%) y se obtuvo la línea celular HeLa PARP-1kd. En estas células, la frecuencia de rupturas cromatídicas y cromosómicas/célula fue de 0,45±0,06 luego del tratamiento con ETO; mientras que en las células HeLa RAD21kd tratadas con DNA-PKi/ETO fue de 3,3±0,2 (p=0,0001). En el caso de cromosomas dicéntricos/célula, en las células HeLa PARP-1kd tratadas con ETO fue de 0,26±0,05 y en las células HeLa RAD21kd tratadas con DNA-PKi/ETO de 1,90±0,13 (p=0,0001).El incremento de las AC observado frente a la falta de las vías de reparación c-NHEJ y HR y su disminución en células deficientes en PARP-1, evidenciaron la contribución de alt-EJ en la formación de estas alteraciones cromosómicas obtenidas.
Además, se analizó el IM en las células HeLa NS, HeLa RAD21kd y HeLa PARP-1kd a las 28 h luego de los tratamientos (Figuras 5e y 5f). Los IM de las células HeLa NS tratadas con DMSO o con el inhibidor de DNAPKcs fueron más altos que los observados en las células HeLa RAD21kd (p<0,05). Mientras que en HeLa NS, el tratamiento con DNA-PKi/ETO provocó una reducción significativa del IM en comparación con el valor obtenido con ETO (p=0,0001), estos tratamientos no causaron cambios notorios en el IM de las células HeLa RAD21kd (Figura 5e).
A su vez, como se muestra en la Figura 5f, los IM obtenidos en las células HeLa PARP-1kd expuestas a DMSO o a ETO no mostraron diferencias estadísticamente significativas. Por lo tanto, la disminución de las AC en las células HeLa PARP-1kd no se debió a un descenso del número de metafases analizables.
Con el fin de evaluar la inestabilidad cromosómica originada por la participación de alt-EJ en la reparación de las RDC inducidas por ETO, se analizaron los MN presentes en células BN a las 96 h (a largo plazo). Los MN son cuerpos citoplasmáticos de naturaleza nuclear, que no son incorporados correctamente a las células hijas durante la división celular y surgen como resultado de un daño directo sobre el ADN (fragmentos acéntricos, clastogenicidad) o por el retraso de cromosomas enteros durante la segregación (aneugenicidad) (Chondrou et al., 2018). La combinación del ensayo de MN con la detección de focos γH2AX en ellos, permite discriminar su origen: los agentes clastogénicos producen MN que contienen focos γH2AX (MN γH2AX+) y los aneugénicos inducen MN que no los presentan (MN γH2AX-). Los datos sobre la presencia de MN en las células HeLa NS y HeLa RAD21kd se muestran en la Figura 6. La mayoría de las células BN de ambas líneas celulares poseían un MN. La proporción entre los MN de las células BN tratadas y aquellos presentes en las células BN controles (tratadas con DMSO) demostró un aumento significativo en las células BN HeLa RAD21kd (p=0,001, Figura 6a). Dentro de los MN obtenidos se observó un importante aumento de MN γH2AX- en las células HeLa NS y HeLa RAD21kd tratadas con DMSO o con el inhibidor de DNA-PKcs (Figuras 6b y 6c). En cambio, el tratamiento con ETO produjo una disminución de MN γH2AX-, incrementándose el porcentaje de los MN γH2AX+ (61,2±6,6% en HeLa NS vs. 53,6±16,8% en HeLa RAD21kd). Los porcentajes de los MN γH2AX+ alcanzaron sus valores más altos (>70%) con el tratamiento combinado DNA-PKi/ ETO en ambas líneas celulares. Las células RAD21kd tratadas con la combinación del inhibidor de DNAPKcs y ETO mostraron un marcado aumento de MN, siendo la mayoría de ellos γH2AX+, lo cual indicó que los fragmentos acéntricos y los cromosomas dicéntricos podrían contribuir a la formación de los MN obtenidos.
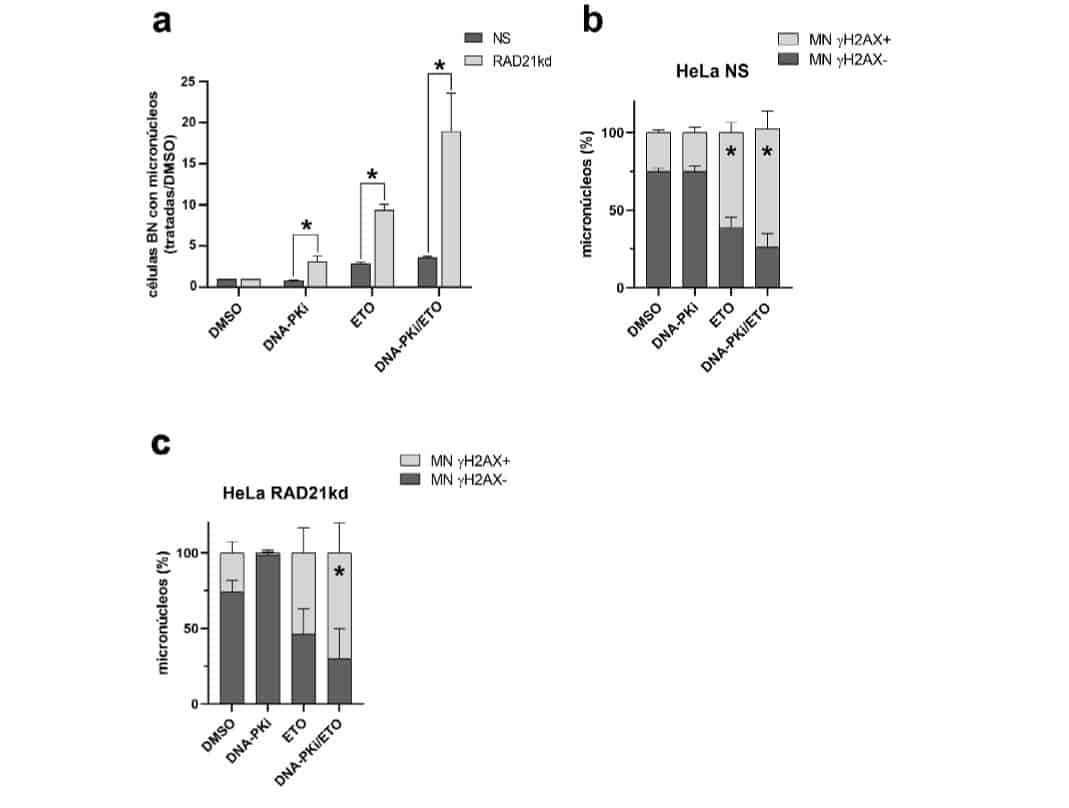
Figura 6. Células binucleadas (BN) con micronúcleos (MN) a las 96 h postratamientos.
a. Proporción entre las células BN con MN luego de los diferentes tratamientos y las células BN con MN tratadas con DMSO (control) en HeLa NS y Hela RAD21kd, *p=0,001; b y c. Porcentajes de MN con (γH2AX+) o sin (γH2AX-) rupturas de doble cadena, *p=0,0001 tratados vs. sus respectivos controles en células HeLa NS y Hela RAD21kd.
DISCUSIÓN
ETO es una droga utilizada en el tratamiento de leucemias, linfomas y tumores sólidos. A pesar de su amplio y efectivo uso, los pacientes tratados con ETO presentan el riesgo potencial de desarrollar leucemias secundarias relacionadas a la terapia, las cuales suelen manifestarse luego de un periodo de latencia corto (Zhang et al., 2021). La incidencia de estas leucemias se incrementó debido a una mayor tasa de sobrevida de los pacientes y al uso de regímenes quimioterapéuticos más intensos para tratar tumores primarios.
Los rearreglos entre cromosomas como las translocaciones, representan las anomalías cariotípicas características de las leucemias (Roukos y Misteli, 2014). En este sentido, la vía de reparación alt-EJ remueve las RDC ante la ausencia de c-NHEJ y de HR, promoviendo un procesado incorrecto de estas lesiones, lo cual origina grandes alteraciones en los sitios de unión de los extremos rotos y la formación de rearreglos cromosómicos (Iliakis et al., 2015). En base a estas observaciones se analizó la presencia de daño cromosómico inducido por ETO en células humanas deficientes en c-NHEJ y HR y su progresión a lo largo de las divisiones celulares.
Las células HeLa deficientes en RAD21 (HeLa RAD21kd) se generaron mediante transfecciones estables con un shRNAmir específico. Nuestros resultados revelaron un porcentaje menor en la expresión de este gen en comparación al valor obtenido (~55-62%) por Xu et al. (2011) en la línea de cáncer de mama MDA-MB-231 utilizando un sistema de silenciamiento génico similar.
La deficiencia en la subunidad RAD21 altera la reparación de las RDC mediante HR y la presencia de ICH es una medida de la activación de la recombinación en células de vertebrados (Sonoda et al., 1999). Los niveles espontáneos de ICH/cromosoma en las células HeLa RAD21kd fueron menores en relación a las células HeLa NS. Similares resultados se obtuvieron en células embrionarias humanas 293/A658 deficientes en RAD21 (SCC1), donde la frecuencia de ICH/metafase fue la mitad de la hallada en células proficientes (Potts et al., 2006).
Cuando se evaluó la sensibilidad de HeLa RAD21kd a ETO se observó que estas células fueron hipersensibles a partir de la concentración más baja empleada en relación a las células HeLa NS. Nuestros resultados están en concordancia con los obtenidos por Atienza et al. (2005), quienes reportaron una mayor sensibilidad de las células de cáncer de mama MCF-7 deficientes en RAD21 cuando fueron tratadas con ETO.
Luego se determinó la presencia de células con RDC (γH2AX+) en la fase G2 del ciclo celular en ambas líneas celulares expuestas a la concentración de ETO elegida, 2 µg/ml. La marcación γH2AX indica una respuesta celular temprana frente al daño inducido en el ADN y es un biomarcador sensible y específico para la detección de RDC (Takahashi y Ohnishi, 2005). En las células HeLa NS y HeLa RAD21kd se obtuvo un marcado aumento en el porcentaje de células con RDC (~90%). En estudios previos, hemos observado la inducción de estas lesiones en la fase G2 de fibroblastos humanos normales (de Campos Nebel et al., 2010) y de células HeLa (Palmitelli et al., 2015) luego del tratamiento con ETO.
La ausencia de la vía HR se consiguió mediante el silenciamiento de RAD21 y la deficiencia de c-NHEJ se logró utilizando un potente inhibidor selectivo de DNA-PKcs, NU7026. Este inhibidor tiene una actividad 60 veces más alta contra DNA-PKcs en relación a otras fosfatidilinositidil 3-quinasas, como ataxia telangiectasia mutada (ATM) y ataxia telangiectasia y rad3-relacionada (ATR) (Willmore el al., 2004). Los resultados exhibieron un aumento sinérgico de rupturas e intercambios de cromátidas frente a ETO en un contexto deficiente en c-NHEJ y en HR en la primera metafase postratamiento.
A su vez, el daño inducido se evaluó en la interfase
G1 posmitótica mediante la cuantificación de núcleos con ≥20 focos γH2AX en células BN. Estos focos pueden ser el resultado de lesiones persistentes sin reparar o incorrectamente reparadas, y/o a las producidas durante la mitosis previa (Ricoul et al., 2019). En consecuencia, los focos γH2AX observados en la interfase G1 posmitótica reflejarían la presencia de rupturas cromatídicas (lesiones sin reparar), de figuras de intercambio de cromátidas (lesiones mal reparadas), y/o de las rupturas de estas figuras de intercambio ocasionadas durante la migración, hacia polos opuestos, de los centrómeros durante anafase.El tratamiento con ETO o con la combinación DNA-PKi/ETO causó un marcado aumento de células BN con ≥20 focos γH2AX en HeLa RAD21kd en comparación a HeLa NS, sin una reducción importante en el porcentaje de células BN.
En las células tumorales HeLa no se observa una acumulación celular en la fase G1 en presencia de un ADN dañado debido a la falta o disminución de la expresión de la proteína p53 (Del Nagro et al., 2014). Es por esto que el análisis del ciclo celular se focalizó en el punto de control G2/M.
A las 8 h se obtuvo una mayor proporción de células HeLa NS en la fase G2/M respecto a las células HeLa RAD21kd. Estos resultados indicaron que, a pesar del mayor daño cromosómico presente en RAD21kd, estas células continuaron su progresión durante el ciclo celular por poseer un punto de control G2/M defectuoso. En este sentido, Watrin y Peters (2006) reportaron que células HeLa deficientes en RAD21 (SCC1) pudieron entrar en mitosis a pesar de la presencia de cromosomas altamente fragmentados luego de la exposición a ETO 5 µM (~3 µg/ml) por 15 min durante la fase G2. Por lo tanto, la subunidad proteica RAD21 promovería la activación del punto de control G2/M frente al daño inducido.
A las 16 h, la acumulación en G2/M disminuyó en las células HeLa NS tratadas con DNA-PKi/ETO, lo cual indicaría el reinicio del ciclo celular. Por otro lado, HeLa RAD21kd presentó una proporción de células en la fase G2/M que se mantuvo desde las 8 h luego de los tratamientos con ETO y aún más con DNA-PKi/ETO. Según datos reportados, los fibroblastos embrionarios de ratón deficientes en RAD21 (RAD21+/-) exhibieron un aumento de células en la fase G2/M a las 24 h luego de exponerlos a radiación ionizante al compararlos con los fibroblastos controles wild-type (Xu et al., 2010).
Nuestros resultados junto a los antecedentes bibliográficos citados, indicarían que la falta de RAD21 llevaría a un arresto celular insuficiente en el punto de control G2/M a tiempos cortos, aunque el porcentaje de las células acumuladas podría mantenerse en el tiempo.
Asimismo, los inhibidores de DNA-PKcs influyen sobre la acumulación celular en G2/M. La distribución de las poblaciones celulares de HeLa NS y HeLa RAD21kd en las diferentes fases mostró un ligero incremento en G2/M en presencia del inhibidor de DNA-PKcs NU7026 en combinación con ETO. En el mismo sentido, las células de leucemia mieloide crónica K562 expuestas a ETO presentaron una mayor acumulación en G2/M en presencia de NU7026 (Willmore et al., 2004). La adición de otro inhibidor de DNA-PKcs, NU7441 aumentó el número de células de carcinoma nasofaríngeo SUNE1 en la fase G2/M luego de la exposición a radiación ionizante (Dong et al., 2018).
A las 28 h, las metafases de la segunda división mitótica revelaron un mayor incremento de AC luego del tratamiento con ETO en un contexto deficiente en c-NHEJ y en HR, en comparación con los valores obtenidos en la primera metafase. Las células HeLa RAD21kd en presencia de DNA-PKi/ETO utilizaron la vía alt-EJ para reparar las RDC persistentes, llevando a un aumento sinérgico de cromosomas dicéntricos. Alt-EJ es una vía lenta que remueve los extremos libres del ADN, muy citotóxicos para el genoma, induciendo al mismo tiempo un aumento de rearreglos cromosómicos (Iliakis et al., 2015). Además, las deficiencias de RAD21 (Gelot et al., 2016) o de DNA-PKcs (Gunn et al., 2011) están asociadas a la aparición de rearreglos al reunir en forma incorrecta los extremos del ADN.
La participación de la vía alt-EJ se confirmó con los valores obtenidos en las células HeLa PARP-1kd. En estas células deficientes en PARP-1, la frecuencia de rupturas cromatídicas y cromosómicas, y de cromosomas dicéntricos se redujo luego del tratamiento con ETO.
Los cromosomas dicéntricos son inductores de inestabilidad cromosómica a través del ciclo de rupturafusión-puente (breakage-fusion-bridge, BFB cycle). El ciclo BFB comienza con la formación de rupturas en el cromosoma que se fusionan originando cromosomas dicéntricos, o cromátidas hermanas unidas, y en anafase estas estructuras crean puentes cromatínicos al dirigirse los dos centrómeros hacia polos opuestos. Los puentes se rompen y los extremos rotos vuelven a fusionarse, repitiendo el ciclo BFB (Gisselsson et al., 2000).
Los MN son estructuras nucleares pequeñas que pueden originarse por fragmentos cromosómicos acéntricos o cromosomas dicéntricos, que se pueden incorporar en el genoma o ser excluidos de las células durante las subsecuentes divisiones celulares. Los MN son considerados indicadores de genotoxicidad y de inestabilidad cromosómica. La inestabilidad cromosómica es una característica de los cánceres humanos y está asociada a un pobre pronóstico, a metástasis y a resistencia terapéutica (Bakhoum y Cantley, 2018). Se han observado MN en tumores sólidos, en neoplasias hematológicas o en linfocitos de sangre periférica de pacientes durante la evolución de un cáncer (Tijhuis et al., 2019; Siri et al., 2021).
A las 96 h, se observó un aumento de la proporción de MN en las células BN HeLa RAD21kd, alcanzando el mayor valor en aquellas tratadas con DNA-PKi/ETO. A su vez, el 70% de estos MN presentó focos γH2AX, resultado de fragmentos acéntricos y/o cromosomas dicéntricos. Las líneas celulares linfoblastoides humanas delecionadas en RAD21 exhibieron un aumento de MN y de puentes nucleoplásmicos luego de la exposición a radiación ionizante (Deardorff et al., 2012). Un importante incremento de MN espontáneos se observó luego del silenciamiento de RAD21 en células epiteliales de cáncer colorectal HCT116 y en fibroblastos humanos inmortalizados hTERT (Leylek et al., 2020). Por otro lado, nuestros resultados previos han señalado un significativo aumento de MN en presencia de DNA-PKi/ ETO en células HeLa (Palmitelli et al., 2015).
Yamauchi et al. (2011) indicaron que los cromosomas dicéntricos y las translocaciones cromosómicas surgen por una reparación inadecuada de las RDC y que ambas aberraciones ocurren por mecanismos idénticos. El análisis de las uniones de los puntos de ruptura (breakpoints) de las translocaciones cromosómicas en tumores humanos reveló características típicas (microhomologías, extensa resección terminal) de la intervención de alt-EJ en la formación de translocaciones (Deriano y Roth, 2013; Patterson-Fortin y D’Andrea, 2020).
En presencia de ETO, alt-EJ generó rearreglos cromosómicos y MN, eventos asociados con el fenotipo de inestabilidad cromosómica, la transformación celular y el posible inicio de un proceso tumorigénico. En base a esto, y a la reducción de cromosomas dicéntricos observada en células deficientes en PARP-1, la vía alt-EJ podría constituirse en un blanco terapéutico promisorio para evitar la aparición de leucemias secundarias en los pacientes tratados con ETO.
FUENTES DE FINANCIACIÓN
Esta investigación fue financiada por el Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) PIP 0114/14, Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) PICT 2015-3049 y Fundación A. J. Roemmers 2017.
AGRADECIMIENTOS
Los autores desean expresar un especial agradecimiento a Mariana Gómez por su contribución en la compaginación de las figuras correspondientes al presente artículo.
CONTRIBUCIÓN DE LOS AUTORES
Jacqueline Kramar (JK), Micaela. Palmitelli (MP), Marcelo de Campos-Nebel (MCN), Marcela GonzálezCid (MGC) contribuyeron con la concepción y el diseño del estudio.
JK, MP, MCN y MGC prepararon el material y realizaron los diferentes experimentos (cultivos celulares, tratamientos químicos, técnicas empleadas para la observación en microscopio óptico o fluorescente, análisis mediante el citómetro de flujo e inmunomarcaciones); así mismo, participaron en la obtención de los resultados y su posterior análisis. Todos los autores contribuyeron en la elaboración del manuscrito, y leyeron y aprobaron la versión final. Los autores declaran no tener ningún conflicto de interés.
BIBLIOGRAFÍA
Atienza J.M., Roth R.B., Rosette C., Smylie K.J., Kammerer S., Rehbock J., Ekblom J., Denissenko M.F. (2005) Suppression of RAD21 gene expression decreases cell growth and enhances cytotoxicity of etoposide and bleomycin in human breast cancer cells. Mol. Cancer Ther. 4: 361-368.
Bakhoum S.F., Cantley L.C. (2018) The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell 174: 1347-1360.
Baldwin E.L., Osheroff N. (2005) Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anticancer Agents 5: 363-372.
Ceccaldi R., Rondinelli B., D’Andrea A.D. (2016) Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell. Biol. 26: 52-64.
Chen T., Sun Y., Ji P., Kopetz S., Zhang W. (2015) Topoisomerase IIα in chromosome instability and personalized cancer therapy. Oncogene 34: 4019-4031.
Cheng H., Zhanga N., Patia D. (2020) Cohesin subunit RAD21: From biology to disease. Gene 758: 144966
Chondrou V., Trochoutsou K., Panayides A., Efthimiou M., Stephanou G., Demopoulos N.A. (2018) Combined study on clastogenic, aneugenic and apoptotic properties of doxorubicin in human cells in vitro. J. Biol. Res. (Thessalon) 25 (1): 1-13.
Deardorff M.A., Wilde J.J., Albrecht M., Dickinson E., Tennstedt S., Braunholz D., Mönnich M., Yan Y., Xu W., Gil-Rodríguez M.C., Clark D., Hakonarson H., Halbach
S., Michelis L.D., Rampuria A., Rossier E., Spranger S., Van Maldergem L., Lynch S.A., Gillessen-Kaesbach G., Lüdecke H.J., Ramsay R. G., McKay M.J., Krantz I.D., Xu H., Horsfield J.A., Kaiser F.J. (2012) RAD21 mutations cause a human cohesinopathy. Am. J. Hum. Genet. 90: 1014-1027.
de Campos-Nebel M., Larripa I., González-Cid M. (2010) Topoisomerase II-mediated DNA damage is differently repaired during the cell cycle by nonhomologous end joining and homologous recombination. PLoS One 5 (9): e12541.
Deckbar D., Birraux J., Krempler A.,
Tchouandong L., Beucher A., Walker S., Stiff T., Jeggo P., Löbrich M. (2007) Chromosome breakage after G2 checkpoint release. J. Cell. Biol. 176: 749-755.
Del Nagro C.J., Choi J., Xiao Y., Rangell L., Mohan S., Pandita A., Zha J., Jackson P.K., O’Brien T. (2014) Chk1 inhibition in p53deficient cell lines drives rapid chromosome fragmentation followed by caspaseindependent cell death. Cell Cycle 13: 303314.
Deriano L., Roth D.B. (2013) Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu. Rev. Genet. 47: 433-455.
De Vos M., Schreiber V., Dantzer F. (2012) The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem. Pharmacol. 84: 137-146.
Dong J., Ren Y., Zhang T., Wang Z., Ling C.C., Li G.C., He F., Wang C., Wen B. (2018)
Inactivation of DNA-PK by knockdown DNAPKcs or NU7441 impairs non-homologous end-joining of radiation-induced double strand break repair. Oncol. Rep. 39: 912-920.
Gelot C., Guirouilh-Barbat J., Le Guen T.,
Dardillac E., Chailleux C., Canitrot Y., Lopez B.S. (2016) The cohesin complex prevents the end joining of distant DNA doublestrand ends. Mol. Cell 61: 15-26.
Gisselsson D., Louise Pettersson L., Höglund M., Heidenblad M., Gorunova L., Wiegant J., Mertens F., Dal Cin P., Mitelman F., Nils Mandahl N. (2000) Chromosomal breakagefusion-bridge events cause genetic intratumor heterogeneity. PNAS 97: 53575362.
Gunn A., Bennardo N., Cheng A., Stark J.M. (2011) Correct end use during end joining of multiple chromosomal double strand breaks is influenced by repair protein RAD50, DNAdependent Protein Kinase DNA-PKcs, and transcription context. J. Biol. Chem. 286: 42470-42482.
Heidinger-Pauli J.M., Unal E., Koshland D. (2009) Distinct targets of the Eco1 acetyltransferase modulate cohesion in S phase and in response to DNA damage. Mol. Cell 34: 311-321.
Iliakis G., Murmann T., Soni A. (2015) Alternative end-joining repair pathways are the ultimate backup for abrogated classical nonhomologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 793: 166-175.
Kopa P., Macieja A., Galita G., Witczak Z.J., Poplawski T. (2019) DNA double strand breaks repair inhibitors: Relevance as potential new anticancer therapeutics. Curr. Med. Chem. 26: 1483-1493.
Leylek T.R., Jeusset L.M., Lichtensztejn Z., McManus K.J. (2020) Reduced expression of genes regulating cohesion induces chromosome instability that may promote cancer and impact patient outcomes. Sci. Rep. 10: 592.
Litwin I., Pilarczyk E., Wysocki R. (2018) The emerging role of cohesin in the DNA damage response. Genes 9 (12): 581.
McKie S.J., Neuman K.C., Maxwell A. (2021) DNA topoisomerases: Advances in understanding of cellular roles and multi-protein complexes via structure-function analysis. BioEssays 43: 2000286.
Mladenov E., Magin S., Soni A., Iliakis G. (2016) DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferationdependent regulation. Semin. Cancer Biol. 37-38: 51-64.
Mohiuddin I.S., Kang M.H. (2019) DNA-PK as an emerging therapeutic target in cancer. Front. Oncol. 9: 635.
Montecucco A., Zanetta F., Biamonti G. (2015) Molecular mechanisms of etoposide. EXCLI J. 14: 95-108.
Obe G., Durante M. (2010) DNA double strand breaks and chromosomal aberrations. Cytogenet. Genome Res. 128: 8-16.
Palmitelli M., de Campos-Nebel M., GonzálezCid M. (2015) Progression of chromosomal damage induced by etoposide in G2 phase in a DNA-PKcs deficient context. Chromosome Res. 23: 719-732.
Patterson-Fortin J., D’Andrea A.D. (2020) Exploiting the microhomology-mediated end-joining pathway in cancer therapy. Cancer Res. 80: 4593-4600.
Peters J-M., Tedeschi A., Schmitz J. (2008) The cohesin complex and its roles in chromosome biology. Genes Dev. 22: 3089-3114.
Pfaffl M.W. (2004) Quantification strategies in real-time PCR. In: Bustin S. A. (Ed.) A-Z of quantitative PCR. International University Line, La Jolla, CA pp. 87-112.
Pierce A.J., Stark J.M., Araujo F.D., Moynahan M.E., Berwick M., Jasin M. (2001) Doublestrand breaks and tumorigenesis. Trends Cell Biol. 11: S52-S59.
Potts P.R., Porteus M.H., Yu H. (2006) Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double-strand breaks. EMBO J. 25: 3377-3388.
Ranjha L., Howard S.M., Cejka P. (2018) Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes. Chromosoma 127: 187-214.
Ricoul M., Gnana Sekaran T.S., Brochard P., Herate C., Sabatier L. (2019) γ-H2AX foci persistence at chromosome break suggests slow and faithful repair phases restoring chromosome integrity. Cancers 11: 1397.
Roukos V., Misteli T. (2014) The biogenesis of chromosome translocations. Nat. Cell. Biol. 16: 293-300.
Sallmyr A., Tomkinson A.E. (2018) Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 293: 10536-10546.
Scully R., Panday A., Elango R., Willis N. A. (2019) DNA double-strand break repairpathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 20: 698-714.
Siri S.O., Martino J., Gottifredi V. (2021) Structural chromosome instability: Types, origins, consequences, and therapeutic opportunities. Cancers 13: 3056.
Sonoda E., Sasaki M.S., Morrison C., Yamaguchi-Iwai Y., Takata M., Takeda S. (1999) Sister chromatid exchanges are mediated by homologous recombination
in vertebrate cells. Mol. Cell Biol. 19: 51665169.
Takahashi A., Ohnishi T. (2005) Does gammaH2AX foci formation depend on the presence of DNA double strand breaks? Cancer Lett. 229: 171-179.
Tijhuis A.E., Johnson S.C., McClelland S.E. (2019) The emerging links between chromosomal instability (CIN), metastasis, inflammation and tumour immunity. Mol. Cytogenet. 12: 17.
Visconti R., Della Monica R., Grieco D. (2016) Cell cycle checkpoint in cancer: a therapeutically targetable double-edged sword. J. Exp. Clin. Cancer Res. 35: 153.
Vítor A.C., Huertas P., Legube G., de Almeida S.F. (2020) Studying DNA double-strand break repair: An ever-growing toolbox. Front. Mol. Biosci. 7: 24.
Wang H., Xu X. (2017) Microhomologymediated end joining: new players join the team. Cell Biosci. 7: 6.
Watrin E., Peters J.M. (2006) The cohesin complex is required for the DNA damageinduced G2/M checkpoint in mammalian cells. The EMBO J. 28: 2625-2635.
Willmore E., de Caux S., Sunter N.J., Tilby M.J., Jackson G.H., Austin C.A., Durkacz B.W. (2004) A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood 103: 4659-4665.
Xu H., Balakrishnan K., Malaterre J., Beasley M., Yan Y., Essers J., Appeldoorn E., Thomaszewski J.M., Vazquez M., Verschoor S., Lavin M.F., Bertonchello I., Ramsay R.G., McKay M.J. (2010) Rad21-cohesin haploinsufficiency impedes DNA repair and enhances gastrointestinal radiosensitivity in mice. PLoS One 5: e12112.
Xu H., Yan M., Patra J., Natrajan R., Yan Y., Swagemakers S., Tomaszewski J.M., Verschoor S., Millar E.K., van der Spek P.,
Reis-Filho J.S., Ramsay R.G., O’Toole S.A., McNeil C.M., Sutherland R.L., McKay M.J., Fox S.B. (2011) Enhanced RAD21 cohesin expression confers poor prognosis and resistance to chemotherapy in high grade luminal, basal and HER2 breast cancers. Breast Cancer Res. 13: R9.
Yamauchi M., Suzuki K., Oka Y., Suzuki M., Kondo H., Yamashita S. (2011) Mode of ATMdependent suppression of chromosome translocation. Biochem. Biophys. Res. Commun. 416: 111-118.
Zhang W., Gou P., Dupret J-M., Chomienne C., Rodrigues-Lima F. (2021) Etoposide, an anticancer drug involved in therapy-related secondary leukemia: Enzymes at play. Transl. Oncol. 14: 101169.